Research: Present & Past
Over the past 9 months, the CRG.BMD team has been involved with the following Rare genetic disease studies. The results, when published, should shed light on these disease in the Indian context
Ongoing projects
Identification of a regional cluster of patients with Treacher Collins syndrome: a rare genetic mandibulofacial dysostoses disorder
Background
Over the past two years, physicians at the KS Hegde Hospital noticed
an unusual number of pediatric patients coming in for cleft palate surgery. These patients
also had other symptoms including deformities of the ears, eyes, cheekbones and chin and
hearing loss in a few patients, characteristic of Treacher Collins Syndrome (TCS). TCS is a
rare (1:50,000 occurrence in Europe) disease which is inherited in 40% of cases in an
autosomal dominant manner. Most patients have mutations in the TCOF1 (Treacle) gene, but
more recently mutations in the POLR1C, POLR1D and EFTUD2 genes have also been implicated.
Most patients have mutations in the TCOF1 gene, the protein product of which is involved in
ribosome assembly in the cell. Based on the clinical findings, we sought to confirm the
clinical diagnosis of TCS in these patients using a genetic approach.
Methods
Blood was drawn from eight consented patients, their parents and other
affected members of the family (a total of 24) and DNA isolated for whole exome sequencing
(WES) analysis. Clinical data was documented for each patient as well.
Results:
Six of the eight patients analyzed demonstrated novel frame shift
mutations in the TCOF1 gene, of which five patients had inherited disease while one had a de
novo mutation. The other two patients who lacked mutations in the TCS causal genes (TCOF1,
POLR1D, POLR1C, EFTUD2) are currently being analyzed for potentially novel TCS gene
mutations, or mutations associated with TCS-like syndromes.
Conclusions:
We have identified a cluster of TCS patients in unrelated families
from the South Karnataka- Northern Kerala region of India. This finding is significant as
very few case reports have been recorded thus far, making this the largest single report of
TCS patients from India to date.
A Case report of Lesch-Nyhan syndrome: a very rare genetic disorder
Background
On her visit to the northern Indian state of Uttar Pradesh, one of the
CRG.BMD team members came across a family with a young boy who had been clinically diagnosed
with Lesch-Nyhan syndrome (LNS): a rare disease first described in 1964 at Johns Hopkins
University in the US. LNS is a very rare condition which affects 1:380,000 live births in
the US and less than 10 patients have been described in India so far. It is an X-linked
disease caused by mutations in the HPRT1 gene which results in deficiency of the
hypoxanthine-guanine phosphoribosyltransferase (HGPRT) enzyme, which in turn leads to a
buildup of uric acid in all organs of the body including in the blood and urine due to
reduced purine utilization. The disease is associated with severe gout, kidney problems and
moderate intellectual disability. A defining feature of this syndrome is the tendency of the
patient to self-mutilate. Thus far, no cure has been found for this condition, although many
patients live to be adults. A genetic confirmation of LHN was therefore required for this
patient to rule out other syndromes with some overlapping symptoms including idiopathic
mental retardation, autism, Tourette syndrome and Cornelia de Lange syndrome
Methods and Expected Results:
Clinical documentation and blood samples have been
collected from the 12 year old patient and his parents. DNA extracted from the samples will
be subjected initially to PCR amplification of the 9 exons of the HPRT1 gene followed by
Sanger sequencing. If no mutation is found in the coding region of the HPRT1 gene, other
more intensive Next Gen Sequencing protocols, WES and later WGS, will be used to identify
the causative gene and variant in this patient, and his mother. We have however discovered
that the mother of the patient has had elevated blood uric acid levels and has suffered
pains for a long time. Given that LNS is an X-linked disorder, mild LNS symptoms in the
mother could predict a mutation in the HPRT1 gene in both the mother (carrier) and in the
patient. This possibility remains to be confirmed.
An L-Leucine-rich diet in the clinical response of a patient with Ghosal hematodiaphyseal dysplasia (GHDD): a bench to bedside story
Case Report:
An 18 month old male child presented with fever, a history of anemia
(Hb: average 5.8g/dL). Besides anemia, his CBC was normal and his peripheral blood smear
indicated normocytic anemia. In addition the young patient presented with progressive pallor
and bowing of upper and lower extremities, mild splenomegaly but no hepatomegaly or
lymphadenopathy was observed. Radiologic findings indicated widening and thickening of long
bones, cortical thickening, coarse trabeculae and endosteal sclerosis. Bone marrow
examination revealed thickened bony trabeculae, fibrotic marrow spaces with entrapped
hematopoietic cells with a predominance of mononuclear cells. Since the diagnosis of this
patient seemed unclear at the time, he was given a packed cell transfusion, treated with
prednisolone and discharged. Subsequently, a homozygous mutation in the TBXAS1 gene was
identified in this patient. While the child was born of non-consanguineous parents, both
parents were found to be heterozygous for this mutation. Based on this genetic data, the boy
was diagnosed with the extremely rare- Ghosal Type Hematodiaphyseal Dysplasia (GHDD; OMIM
231095),
Background:
GHDD was first described by Ghoshal about three decades ago in
Kolkata, India where five patients were described with diaphyseal dysplasia and refractory
anemia. It has been described as a rare autosomal recessive disease caused by mutations in
the TBXAS1 gene leading to deficiency of Thromboxane-A synthase which is involved in the
synthesis of Thromboxane A2. It has been suggested that decreased Thromboxane-A synthetase
leads to elevated levels of prostaglandin E2, which likely suppresses erythroid precursors
in the bone marrow, resulting in refractory anemia. GHDD patients have been treated with
blood transfusions and also with low-dose corticosteroids. While most patients respond well
to steroid treatment, it cannot be used in the long-term. Previous studies from our team and
others had shown that the amino acid L-Leucine alleviates both anemia and developmental
defects in a both humans and in a zebrafish model of another bone marrow failure syndrome:
Diamond Blackfan anemia (DBA). Based on these studies, the patient was put on a high Leucine
diet
Treatment:
Over a period of six months the patient was weaned off steroids in a
stepwise manner. His diet was supplemented with 25g of Milk powder (Nestles Everyday) twice
a day ( ~1.8g L-Leucine/day). Changes in Hb levels and growth parameters were measured every
three months
Outcomes and conclusions:
The GHDD patient responded well to a Leucine- rich diet
as measured by normalization of Hb (5.8 to a steady 11.0g/dl) and bone structure over six
years, with no steroids supplementation. The child now leads a normal life of a healthy 12
year old!
Past Research
Studies over the past several years have demonstrated the knowledge and expertise that both co-founders have gained in the areas of Rare genetic diseases and bone marrow failureforma syndromes. A selected list of publications attest to these achievements.
Consortium Publications
- Bhat S, Mittal B, Nanjappa DP, Chakraborty G, Shetty S, Khanna-Gupta A, Chakraborty A. 2022. Identification of a novel TNFRSF11A gene variant in a rare case of familial osteopetrosis autosomal recessive type 7. Gene Reports 29:101683.

- Shenoy RD, Shetty V, Dheedene A, Menten B, Nanjappa DP, Chakraborty G, Sips P, de Paepe A, Callewaert B, Chakraborty A. 2022. Phenotypic and molecular heterogeneity in Mandibulofacial Dysostoses: A case series from India. The Cleft Palate-Craniofacial Journal 59: 1346-1351

- Bharti Mittal, Dechamma Pandyanda Nanjappa, Gunimala Chakraborty, Anirban Chakraborty, Arati Khanna-Gupta. 2021. A patient with Lesch-Nyhan Syndrome presenting with anesthetic challenges: not an exception, but the rule. Gene Reports 24:101289

Past Publications
Anirban Chakraborty, PhD
- Alexis H Bennett, Marie Francoise O’Donohue, Stacey R Gundry, Aye T Chan, Jeffery Widrick, Isabelle Draper, Anirban Chakraborty, Yi Zhou, Leonard I Zon, Pierre-Emmanuel Gleizes, Alan H Beggs, and Vandana Gupta. 2018. RNA helicase, DDX27 regulates skeletal muscle growth and regeneration by modulation of translational processes. PLoS Genetics 14(3): e1007226
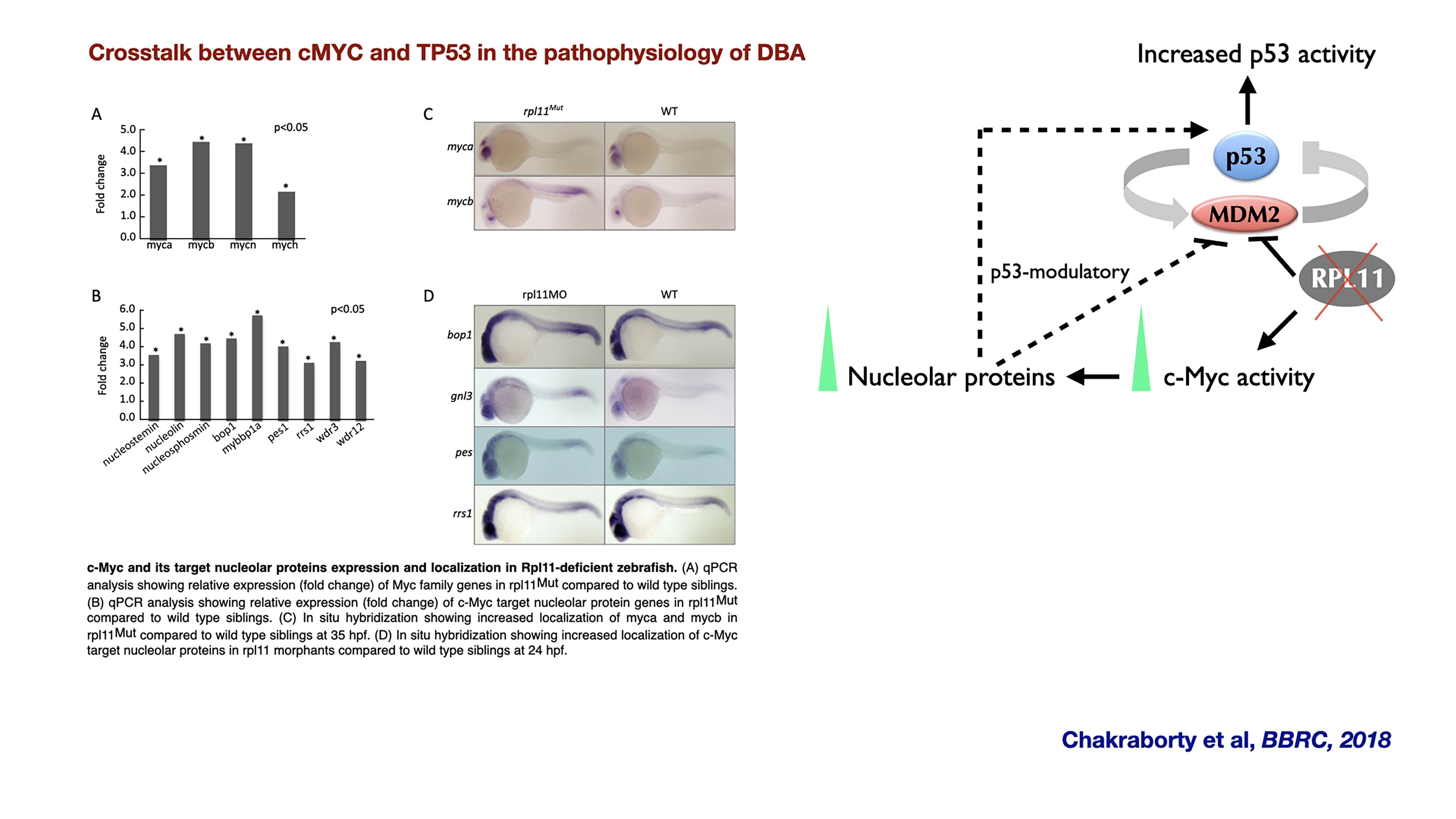
- Anirban Chakraborty, Tamayo Uechi, Yukari Nakajima, Hanna T. Gazda, Marie-Françoise O’Donohue, Pierre-Emmanuel Gleizes, and Naoya Kenmochi. 2018. Cross talk between TP53 and c-Myc in the pathophysiology of Diamond-Blackfan anemia: Evidence from RPL11-deficient in vivo and in vitro models. Biochemical and Biophysical Research Communications 495: 1839- 1845.
- Anirban Chakraborty and Indrani Karunasagar. 2015. Faulty ribosomes and human diseases: mistakes in “assembly line” going unnoticed? Nitte University Journal of Health Science
- Henras A., Plisson-Chastang C., O'Donohue M-F, Anirban Chakraborty, Gleizes P-E. 2014. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdisciplinary Reviews: RNA, 6: 225-242
- G. V. Yadav, Anirban Chakraborty, T. Uechi, and N. Kenmochi. 2014. Ribosomal protein deficiency causes Tp53-independent erythropoiesis failure in zebrafish. The International Journal of Biochemistry & Cell Biology 49: 1-7.
- Anirban Chakraborty and N. Kenmochi. 2012. Ribosomes and Ribosomal Proteins: More than just ‘housekeeping’. eLS. John Wiley and Sons Ltd, Chichester. http://www.els.net/ [DOI: 10.1002/9780470015902.a0005055.pub2]
- S. Higa-Nakamine, T. Suzuki, T. Uechi, Anirban Chakraborty, Y. Nakajima, M. Nakamura, N. Hirano, T. Suzuki, and N. Kenmochi. 2012. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Research 40:391-398.
- Anirban Chakraborty, T. Uechi, and N. Kenmochi. 2011. Guarding the Translation Apparatus: Defective Ribosome Biogenesis and the p53 Signaling pathway. Wiley Interdisciplinary Reviews: RNA, 2: 507-522.
- H. Torihara, T. Uechi, Anirban Chakraborty, M. Shinya, N. Sakai, and N. Kenmochi. 2011. Erythropoiesis failure due to RPS19 deficiency is independent of an activated p53 response in a zebrafish model of Diamond-Blackfan anaemia. British Journal of Hematology 152:648-654.
- Anirban Chakraborty, T. Uechi, S. Higa, H. Torihara and N. Kenmochi. 2009. Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS ONE 4(1): e4152.
- T. Uechi, Y. Nakajima, Anirban Chakraborty, H. Torihara, S. Higa and N. Kenmochi. 2008. Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Human Molecular Genetics 17: 3204-3211.
- T. Uechi, Y. Nakajima, A. Nakao, H. Torihara, Anirban Chakraborty, K. Inoue, and N. Kenmochi 2006. Ribosomal Protein Gene Knockdown Causes Developmental Defects in Zebrafish. PLoS ONE 1: e37.
Arati Khanna-Gupta, PhD
- Khanna-Gupta A, Berliner N. Vitamin B3 boosts neutrophil counts. Nat Med. 2009 Feb;15(2):139-41. doi: 10.1038/nm0209-139.PMID: 19197286
- Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna-Gupta A, Berliner N, Kutok JL, Ebert BL. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117(9):2567-76 (Plenary Paper)
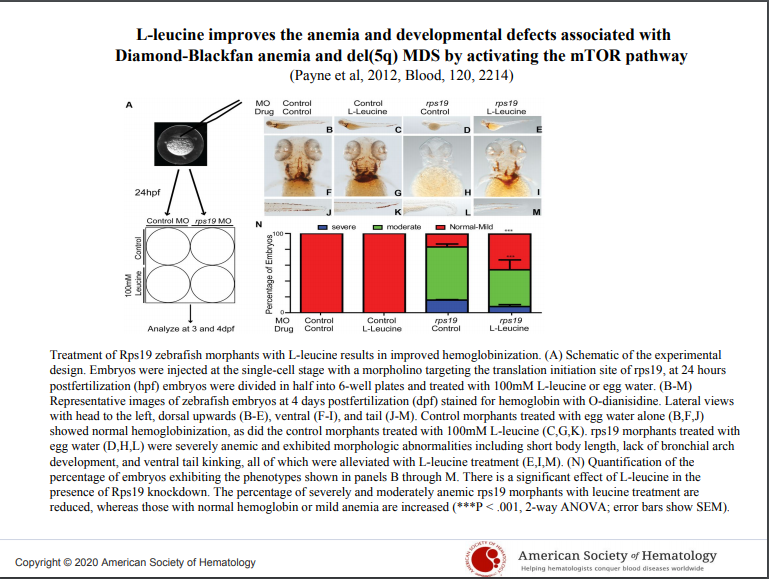
- Payne E, Virgilio M, Narla A, Sun H, Levine M, Paw BH, Look AT, Ebert B, Khanna-Gupta, A Treatment of L-Leucine in both in vivo and in vitro models of ribosomopathies (Diamond Blackfan Anemia (DBA) and 5q- syndrome ((del) 5q) results in an improvement of anemia and developmental defects: evidence for a common pathway. 2012. Blood. 120, 2214-24
- Narla, A, Abayasekara N, Payne E, Hurst SL, Look AT, Berliner N, Ebert BL, Khanna-Gupta, A. L-Leucine improves the anemia of Diamond Blackfan Anemia and 5q- syndrome in a p53- independent way. 2014. Br. J Haematol. 2014
- Abayasekara S and Khanna-Gupta, A Translation Control in Myeloid Disease. In Lawrie CH (ed) Hematology- Science and Practice. 2012. ISBN 979-953-307-516-6 30.
- Khanna-Gupta, A. Bone Marrow failure syndromes: the Ribosomopahies. Journal of Bone Marrow Research. 2013. 1: 106 2013. 31.
- Narla, A, Abayasekara N, Payne E, Hurst SL, Look AT, Berliner N, Ebert BL, Khanna-Gupta, A. L-Leucine improves the anemia of Diamond Blackfan Anemia and 5q- syndrome in a p53- independent way. 2014. Br. J Haematol. 2014
- Shalini H Kumar, Kalpana A, Manikandan Suraj, Mary Shoba Das C, Aparna M, Divya J, Jaicy A, Priya K, Ragav Krishna, Arati Khanna-Gupta and Lakshmi BR Comprehensive genetic analysis of 961 unrelated Duchenne Muscular dystrophy patients: focus on diagnosis, prevention and therapeutic possibilities PLOS ONE. 2020, Jun 19;15(6):e0232654. doi: 10.1371/journal.pone.0232654.eCollection 2020.PMID: 32559196
Bringing together clinicians and researchers under one platform to improve clinical therapies and outcomes for the patients.